Telomeres. Apparently that’s the new buzz word in cosmetics . They come in different sizes – as we age they shorten. It’s been suggested that we could lengthen them to cancel the effects of ageing, or shorten them to cure cancer. But what are they?
They’re an integral part of our chromosomes. The 46 long strings of genes in each human cell are folded up to form chromosomes, which we can see down the microscope. The telomeres are at both ends of each chromosome. They protect the ends of the chromosomes, and stop the chromosomes from sticking to each other. Chromosomes joining together can be a cause of cancer – more about how that works when we look at the promised breakage-fusion-bridge clay model (in stop motion hopefully!).

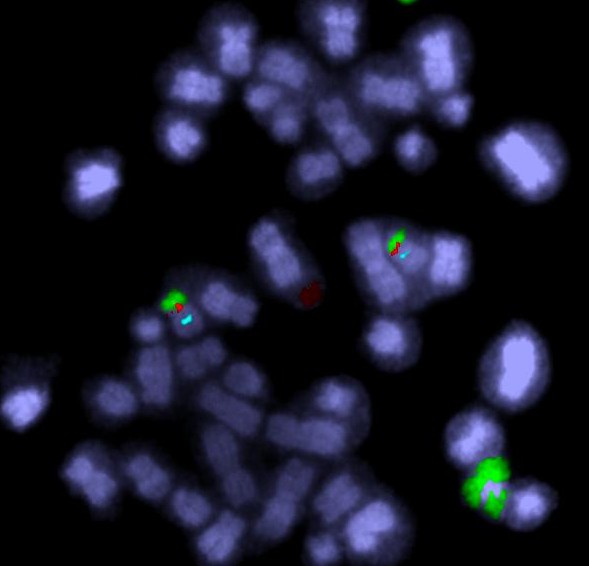
The green spots mark the telomeres on the chromosomes from a leukaemia cell. Spot the two abnormal “ring” chromosomes – no ends, no telomeres (answer next time).
As we age our telomeres get shorter. Telomere shortening has also been associated with other factors such as extreme psychological stress and toxins, including chemotherapy. As well as cancer, short telomeres have been associated with diabetes, cardiovascular disease, osteoarthritis and other diseases. But it also seems that measures like reducing stress, improving diet and exercise may stop or even reverse this premature telomere shortening.
Here’s the paradox – short telomeres can help trigger cancer, but once established the cancer switches on a telomere-lengthening mechanism (usually an enzyme called telomerase) to survive.
And so different researchers are trying contrasting approaches to manipulating telomeres for improved health.
On the one hand, some researchers are looking at the possibility of using the telomere-activating enzyme telomerase to reverse the effects of ageing. Some cosmetics are already available that contain a chemical that’s been reported to activate telomerase. This same chemical is being tested for use as a treatment for some diseases associated with ageing and short telomeres.
On the other hand, because cancers need telomerase to be able to divide indefinitely, other researchers are looking into the possibility of destroying telomerase as a cancer treatment.
These potential treatments will need extensive testing to see if they work and make sure they don’t have unwanted side effects.
From the search for eternal youth to understanding and curing cancer, we haven’t heard the end of telomeres.
INTERESTED IN MORE DETAIL?
A single fertilised egg cell becomes a mature human by growing and dividing into two, many times over. Each time a chromosome makes a copy of itself the telomeres lose a little bit off their ends. The older we get the shorter our telomeres become, so there’s a limited number of times a cell can divide.
That is, unless an enzyme called telomerase is turned on. This enzyme lengthens the telomeres. If you think about it, although most cells in our bodies are programmed to divide a limited number of times, some cells have been dividing for millenia – germ cells – eggs and sperm. It’s cells like these that need telomerase.
Most cancer cells are also able to divide indefinitely by switching on telomerase.
Once the telomeres are dangerously short the chromosomes can start sticking together and becoming abnormal. This stage is called crisis. The cells don’t function properly, and are a cancer risk, and they stop dividing or self-destruct. There are “tumour suppressors” that look after this self-destruction, but occasionally a cell will bypass this (for example by having a mutated tumour suppressor gene), survive and divide. If the cells divide, these abnormal chromosomes can become more abnormal and turn on cancer genes or lose tumour suppressor genes.
HOW MY RESEARCH FITS IN
This process has mostly been studied in lab animals or cells grown in the laboratory in artificial conditions. So we can extrapolate and suggest that chromosomes with short telomeres can join together and cause cancer. Unfortunately chemotherapy is associated with shortened telomeres, and is a risk factor for leukaemia. These therapy-related leukaemias are usually marked by very abnormal chromosomes. In my own research I’ve identified some abnormal leukaemia chromosomes that have been made by chromosomes joining together at the telomeres. This was done by identifying which chromosomes are joined together, AND by looking at the molecular content of the chromosomes. End-to-end joining of the chromosomes is actually a lot more common than it’s thought, at least for the abnormal chromosomes I’ve looked at. The similarity between risk factors for very abnormal leukaemia chromosomes and shortened telomeres is interesting.
One thing I’d like to do is find out how common this joining together of the chromosome ends is, in other types of abnormal chromosomes in leukaemia (AML), and eventually look at other cancers. It would help understand how these cancers are caused and possibly identify ways to prevent this. Other information from this type of study could identify more genes with a role in cancer. This opens up new possibilities for developing treatments.
“Abnormal chromosomes made by the end-to-end joining of two chromosomes….” – that sounds like a segue into the breakage-fusion-bridge cycle. More on that later.
FURTHER READING
E. Blackburn and E. Epel 2012. Too toxic to ignore. Nature 490:169-171 (about stress, disease and telomere shortening). Note, Elizabeth Blackburn is Australia’s only female Nobel Prize winner (in science at least) – she shared the prize for Physiology or Medicine in 2009 for her discovery of telomeres.
C. Buseman 2012. Is telomerase a viable target in cancer? Mutation Research 730:90-97
E. Callaway 2010. Telomerase reverses ageing process. Dramatic rejuvenation of prematurely aged mice hints at potential therapy. Nature 28th November 2010 (published online).
B. de Jesus et al. 2013. Telomerase at the intersection of cancer and aging. Trends in Genetics (available online 19th July 2013)
C. Harley et al 2011. A natural product telomerase activator as part of a health maintenance program. Rejuvenation Research 14:45-56.
R. MacKinnon and L. Campbell 2011. The role of dicentric chromosome formation and secondary centromere deletion in the evolution of myeloid malignancy. Genetics Research International Article ID 643628
R. MacKinnon et al 2011. Unbalanced translocation of 20q in AML and MDS often involves interstitial rather than terminal deletion of 20q. Cancer Genetics 204:153-161.
T. Morin. http://www.dayspamagazine.com/article/spa-products-tale-telomeres A balanced article on telomeres in Dayspa Magazine online.