The Leukaemia Foundation of Australia’s National MDS Day has just passed (14th July… but I was busy eating croissants so this post is a little late).
This time I thought I would tell you about a discovery that was made with the help of MDS.
How do healthy cells turn cancerous? Their DNA gradually accumulates errors. Most of these errors aren’t important, but occasionally they stop the cell from working properly. They might cause a cell to grow out of control – and this can lead to cancer.
Myelodysplastic syndromes, or MDS, are a range of blood disorders caused by such errors in the genes. Some types of MDS are relatively mild, but about a third go on to become acute myeloid leukaemia (AML). Thanks to research on MDS we understand its causes a lot better than we did ten or fifteen years ago.
My lab recently published a paper describing three cases of poor prognosis MDS and one case of AML with unusual but remarkably similar changes to the DNA. This complicated structure could not have been predicted by the standard methods of analysing cancer DNA or chromosomes. These features showed us the likely steps that led to these diseases.
Each long string of DNA is folded up neatly to make a chromosome. This is a Claymation that shows how Barbara McClintock’s classic breakage-fusion-bridge cycle causes chromosome abnormalities. The video shows one way that chromosomes (packages of DNA) can become disorganised.
The telomeres (that cap and protect the ends of the chromosomes) are shown falling off, making sticky chromosome ends which join together (see NOTE 2). It’s well accepted that these changes greatly increase the chance of cancerous gene changes. This process has reproduced many, many times in the lab. The problem is that it’s not often been demonstrated in actual cancers. But we did that.
Sometimes only part of the telomere erodes away – enough is lost that it no longer protects the chromosomes from sticking together. But there can be enough telomere DNA left to be a molecular signature of the telomere.
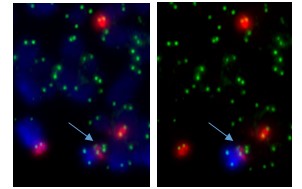
The arrow points to green dots in the middle of a chromosome. This is the left-over telomere signature that tells us that this abnormal chromosome was made by the joining together of sticky chromosome ends that had their telomeres eroded away. The other green dots are at the chromosome ends. The left and right photos show the same cell but in the right one the abnormal chromosome is identified by its red and blue label.
In our four cases we found that there was a small but non-functional piece of telomere DNA left behind where the two chromosomes joined. Because the telomeres didn’t function, the two chromosome ends could stick together. These caused breakage-fusion-bridge events that caused a protective tumour suppressor gene to be lost, and may have also caused cancer-causing genes to multiply.
MDS and AML have similar genetic causes, so if we learn about the causes of one of them it can help us understand the other. This is often the case with cancer research in a broader sense – if we understand the basic mechanisms in one cancer it can help us understand the mechanisms at work in other cancers better. Telomere fusion could potentially play a role in any cancer, so our MDS research is relevant to cancer research in general.
NOTES
- The paper: The dicentric chromosome dic(20;22) is a recurrent abnormality in myelodysplastic syndromes and is a product of telomere fusion. Ruth MacKinnon, Hendrika Duivenvoorden, Lynda Campbell and Meaghan Wall, 2016. Cytogenetic and Genome Research 150(3-4):262-272
- The gene errors discussed here usually occur in the body cells rather than the reproductive cells, so they’re not inherited.
- For simplicity the Claymation shows telomere fusion in chromosomes that are dividing. In fact it probably occurs when the DNA is unravelled in the interphase nucleus.
- This is cross-posted to Fireside Science on the SciFund Challenge network.















