Barbara McClintock published a paper describing the breakage-fusion-bridge (BFB) cycle in 1939. Many of her ideas were well before their time. Like many such profound leaps in thinking, the BFB cycle took a long time to catch on. She wrote in 1973, “I stopped publishing detailed reports long ago when I realized, and acutely, the extent of disinterest and lack of confidence in the conclusions I was drawing …One must await the right time for conceptual change.” Her work was appreciated much later and she was awarded a Nobel Prize in 1983 for her discovery of “jumping genes“.
A few weeks back I introduced this post by describing normal chromosome division. This time we’ll look at the breakage-fusion-bridge cycle. This is one way chromosome division can go wrong. Very wrong, in the sense that it can cause the chromosomes to keep changing, and this can cause cancer.
A human chromosome with two centromeres is abnormal. Chromosomes with two centromeres are not unusual in cancer cells. In fact they’re probably a lot more common than we think, because in both research and diagnostic labs the centromeres are usually not looked at.
To recap, a normal chromosome has one centromere. Before the chromosome divides, the two identical halves (chromatids) are held together at the centromere. When the chromosome divides the centromere splits into two halves, the chromatids become the new chromosomes, and the centromeres take the two new chromosomes in different directions into the two new daughter cells.
So what happens if there are two centromeres? If they’re both aligned so that they head in the same direction it’s not a problem – together they take a complete new chromosome with them. The closer the centromeres are together the more likely this is.
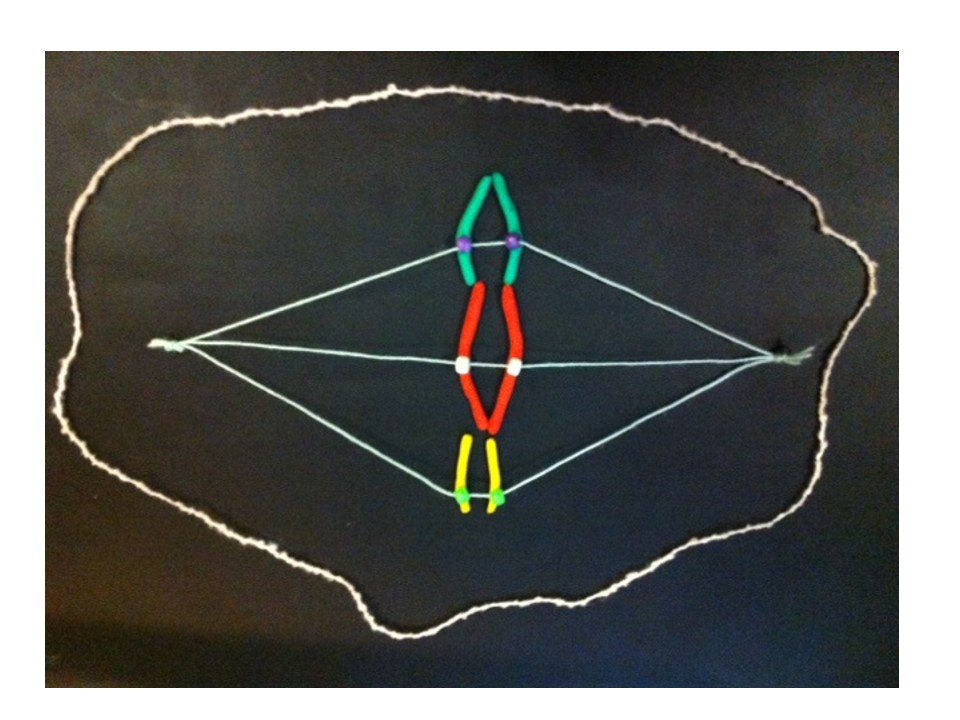
Now follow the pictures and their captions. These describe chromosome division in an abnormal chromosome with two centromeres. Especially follow the yellow dots.

If the two centromeres on a chromosome go in the same direction there’s no problem. But if there’s a twist between the two centromeres when the chromosomes align ready for chromosome division….

….then, when the two halves of each centromere separate they go in opposite directions. We have a “bridge” spanning the gap between the two centromeres.

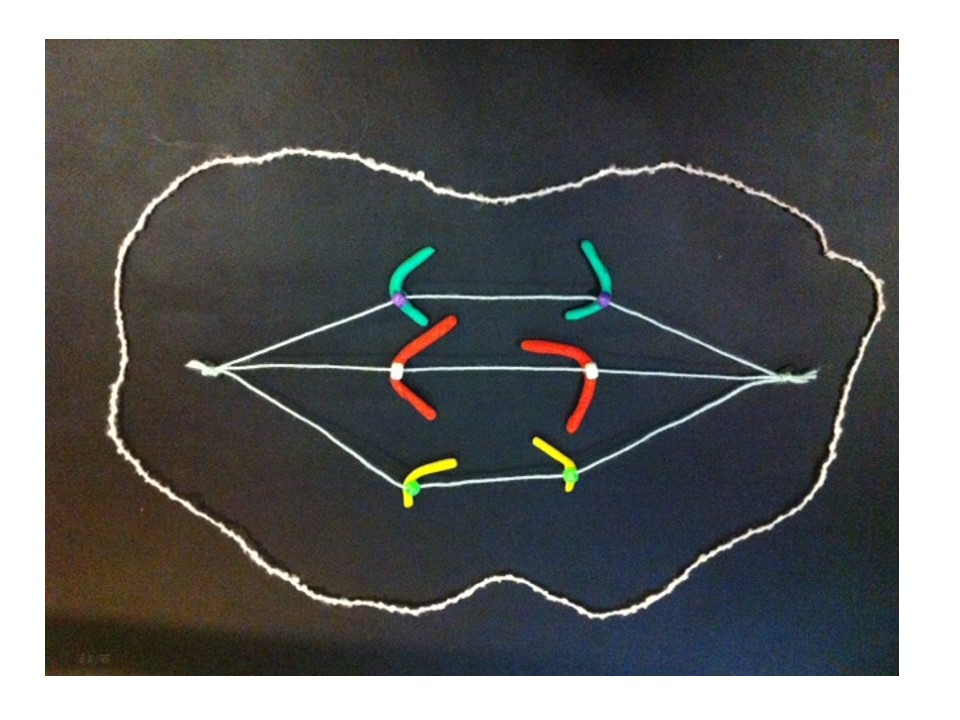
The bridge is stretched and can break.

The broken chromosomes in the new cell join together – the top daughter cell gets an extra copy of the yellow gene. The bottom cell loses this copy of the yellow gene.

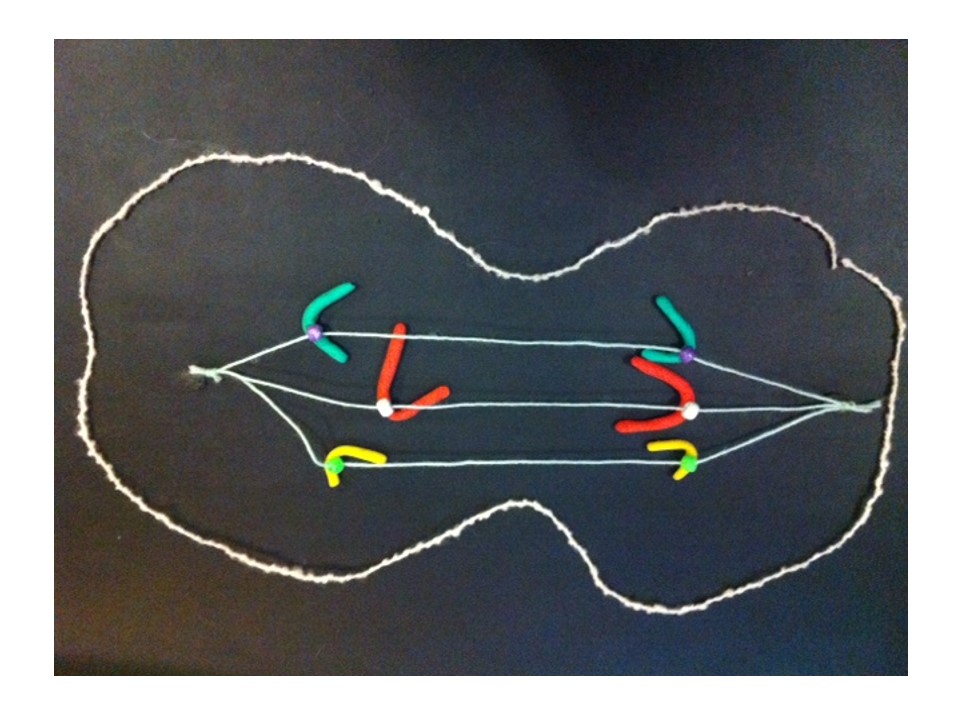
The new chromosome copies itself to make two equal halves.

If this process repeats..

…

Fusion of the broken pieces creates a chromosome with four copies of the yellow gene.

After replication.
If the yellow gene in the pictures is a cancer gene (“oncogene”) the cell with extra copies might grow and multiply faster than its neighbours. We call this natural selection – the cells that can grow faster than their neighbours become more common which means the genetic change causing that is undergoing “positive selection”. Yes, the cells in our body can evolve and we know this best as cancer.
All this change happens between the two centromeres where the bridge forms. So if we find a chromosome with this type of change on one side of the centromere only it’s a clue that this might have been caused by the breakage-fusion-bridge cycle.
These are modelling clay images from my breakage-fusion-bridge claymation. They’re a bit rough but I hope it helps you understand what happens. Many, perhaps most, images demonstrating the BFB cycle show a different version – where the abnormal chromosome is created by two chromatids of one chromosome breaking and joining together. Most examples don’t show the version I’ve presented – where two different chromosomes have joined together. Check this out on Google Images (search for breakage-fusion-bridge).
Here’s the answer to the quiz from the telomere post. The arrows point to the ring chromosomes. Being rings they have no ends, so no telomeres.

Further Reading:
B. McClintock 1939. The Behavior in Successive Nuclear Divisions of a Chromosome Broken at Meiosis. Proc Natl Acad Sci U S A. 1939 August; 25(8): 405–416.
M. Kinsella and V. Bafna 2012. Combinatorics of the Breakage-Fusion-Bridge Mechanism. J Comput Biol. 2012 June; 19(6): 662–678.
R. MacKinnon and L. Campbell 2011. The Role of Dicentric Chromosome Formation and Secondary Centromere Deletion in the Evolution of Myeloid Malignancy. Genetics Research InternationalVolume 2011 (2011), Article ID 643628.
Related articles
- What is a Centromere and How Does it Contribute to Mitosis? (biology.answers.com)
- Understanding the Anatomy of Sister Chromatids (biology.answers.com)